Single-molecule Sequencing
Nanopore sequencing is a third generation technology for nucleic acid sequencing. At its core, this platform consists of a protein Nanopore embedded in an electrically resistant synthetic membrane; a voltage applied across this membrane drives an ionic current through the pore. When a DNA or RNA molecule is ratcheted through the pore, the flow of ions is perturbed in a characteristic way depending the chemical composition of the molecule inside the pore. Thanks to an array of sensors these current changes are recorded and can be analysed to reconstruct the sequence of the DNA or RNA that passed through the pore. Typical sequencing cartridges (flowcells) have hundreds to thousands of nanopores that allow sequencing a large number of molecules in parallel a
nd in real time. Compared to sequencing technologies of previous generations, Nanopore sequencing offers remarkable advantages, e.g. it can read contiguous sequences of length up to the megabase range, it permits sequencing native DNA/RNA molecules without amplification or retrotranscription, it allows the detection of modified bases such as 5-Methylcytosine in DNA or N6-Methyladenosine in RNA. Thanks to these and other advantages, Nanopore sequencing found a broad spectrum of applications, ranging from the generation of highly contiguous genome assemblies, to the identification and phasing of sequence variants to the detection and characterisation of full-length transcript isoforms.
CURRENT PROJECTS
Detection of RNA modifications at single molecule resolution Researchers involved: Longan Mulroney. Project lead: Leonardi
Expression profiling of retrotransposons Researchers involved: Mounika Boddireddy, Camilla Ugolini. Project lead: Leonardi
Profiling of DNA replication dynamics in mammalian cells Researchers involved: Laura Curti, Ottavio Croce. Project lead: Campaner
Characterisation of epi-transcriptional dynamics in cancer Researchers involved: Mattia Furlan, Lucia Coscujuela, iris Tanaka. Project lead: Pelizzola
Profiling of lncRNAs in triple negative breast cancer Researchers involved: Camilla Ugolini, Tommaso Leonardi. Project lead: Nicassio
Characterisation of the SARS-CoV-2 transcriptome Researchers involved: Camilla Ugolini, Logan Mulroney. Project lead: Leonardi
Functional Genomics
 CELL TRACKING the dynamic behaviour of heterogeneous cell populations can be studied by cell tracking, an approach where each cell is labelled with an individual barcode integrated in the cell genome, allowing the analysis of the evolutionary trajectory of the cell populations in different conditions or after treatment. Cell tracking was originally performed by introducing a DNA barcode, which was then recovered analysing the genomic DNA of the cell population in bulk. Recently also RNA barcoding strategies has been created, leading to the possibility of integrating cell tracking with single cell transcriptomics. This technological advancement enabled the possibility of studying the clonal evolution (i.e. “which clones survive treatment?”) with the transcriptional information (i.e. “what is the gene expression profile of the clones surviving treatment?”).
CELL TRACKING the dynamic behaviour of heterogeneous cell populations can be studied by cell tracking, an approach where each cell is labelled with an individual barcode integrated in the cell genome, allowing the analysis of the evolutionary trajectory of the cell populations in different conditions or after treatment. Cell tracking was originally performed by introducing a DNA barcode, which was then recovered analysing the genomic DNA of the cell population in bulk. Recently also RNA barcoding strategies has been created, leading to the possibility of integrating cell tracking with single cell transcriptomics. This technological advancement enabled the possibility of studying the clonal evolution (i.e. “which clones survive treatment?”) with the transcriptional information (i.e. “what is the gene expression profile of the clones surviving treatment?”).
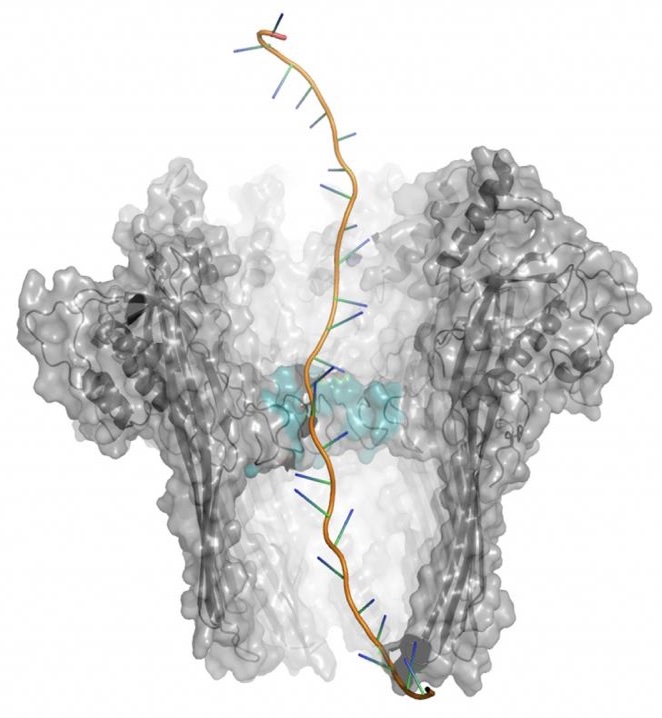
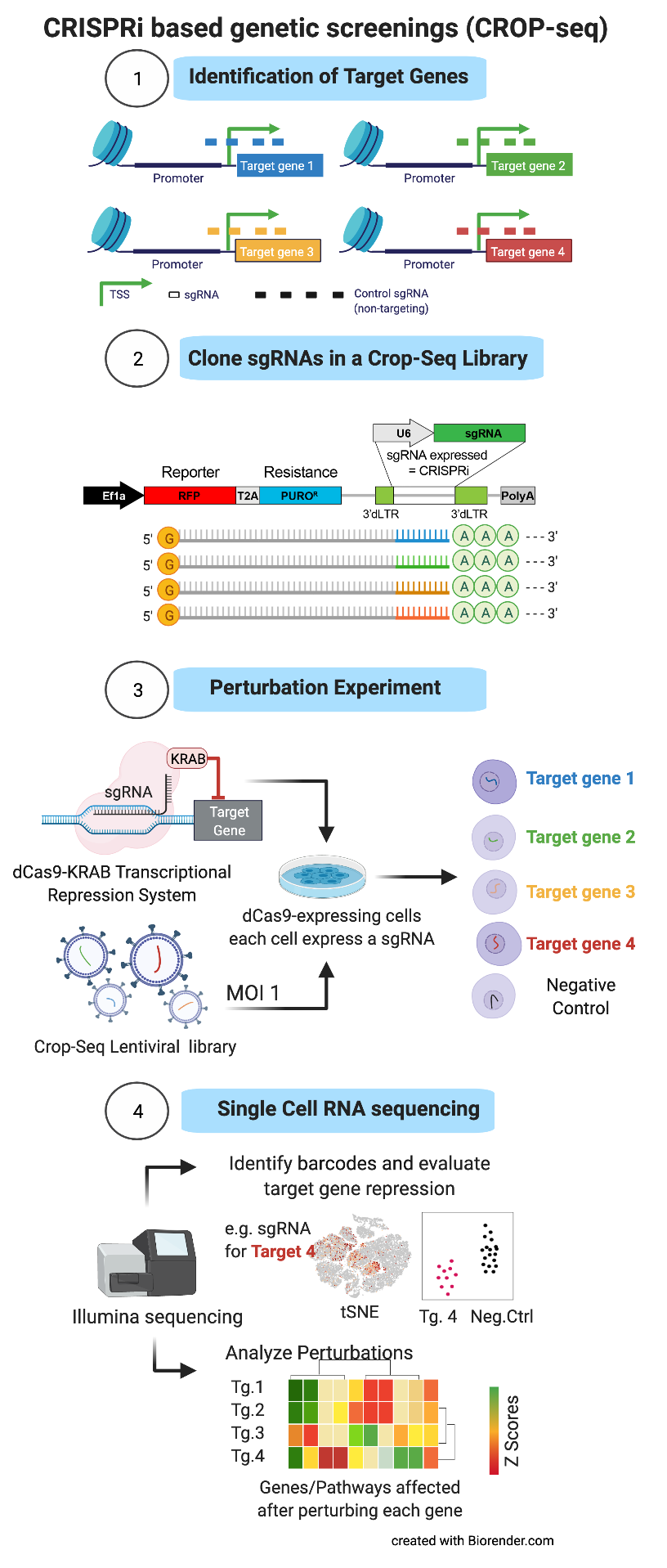
CRISPR-BASED POOLED COMPETITION SCREENINGS The identification of functionally active genetic elements can be performed by CRISPR-based phenotype screenings, an approach where genes or regulatory elements of interest are inactivated by means the so called CRISPR interference approach. The method works by directing specifically at the targets a dCas9-KRAB fusion protein. The dCas9 is required to recognize the region to be inactivated, by means of single guide RNAs (sgRNA) designed as to be complementary to the target DNA sequence of interest. Then the KRAB domain proceed to repress the transcription of the locus, which is thus inactivated. It is possible to study multiple genes at once, by cloning a library of sgRNAs in a lentiviral vector and infecting a cell population introducing a single sgRNA per cell. This complex cell population can be used to perform so-called “pooled competition screens”: genetic perturbations that promote the phenotype of interest will lead to an increase of the sgRNA frequency in the population over time (“drop-in”), whereas perturbations that repress the phenotype will lead to a decrease (“drop-out”).
CRISPR-BASED SINGLE CELL PERTURBATION SCREENING While the above-described phenotype screenings are very informative of the genetic elements that are important for a given biological process, they do not provide any insight into the molecular mechanism underneath. Recently, CRISPR interference has been adapted to single cell RNA sequencing (the so-called CROP-sequencing approach), coupling sgRNAs with a unique transcribed RNA barcode. In this way single-cell RNA-seq profiles are used to investigate the cell programs affected by each individual perturbation (transcriptional fingerprinting), identifying cis- and -trans regulatory networks that control the phenotype of interest.
CURRENT PROJECTS
Single Cell tracking of the tumor initiating process in triple negative breast cancer Researchers involved: Fuentes, Marzi. Project lead: Nicassio
Single Cell characterisation of chemo-adaptation in triple negative breast cancer Researchers involved: Nadalin, Priscazzi, Marzi. Project lead: Nicassio
Functional and molecular characterization of enhancers region associated to cancer genes in breast cancer cells Researchers involved: Giuliani, Marzi. Project lead: Nicassio
Non coding RNAs as critical regulators of the epigenetic and transcriptional plasticity of breast cancer cells Researchers involved: Giuliani, Marzi. Project lead: Nicassio
Single-Cell Genomics
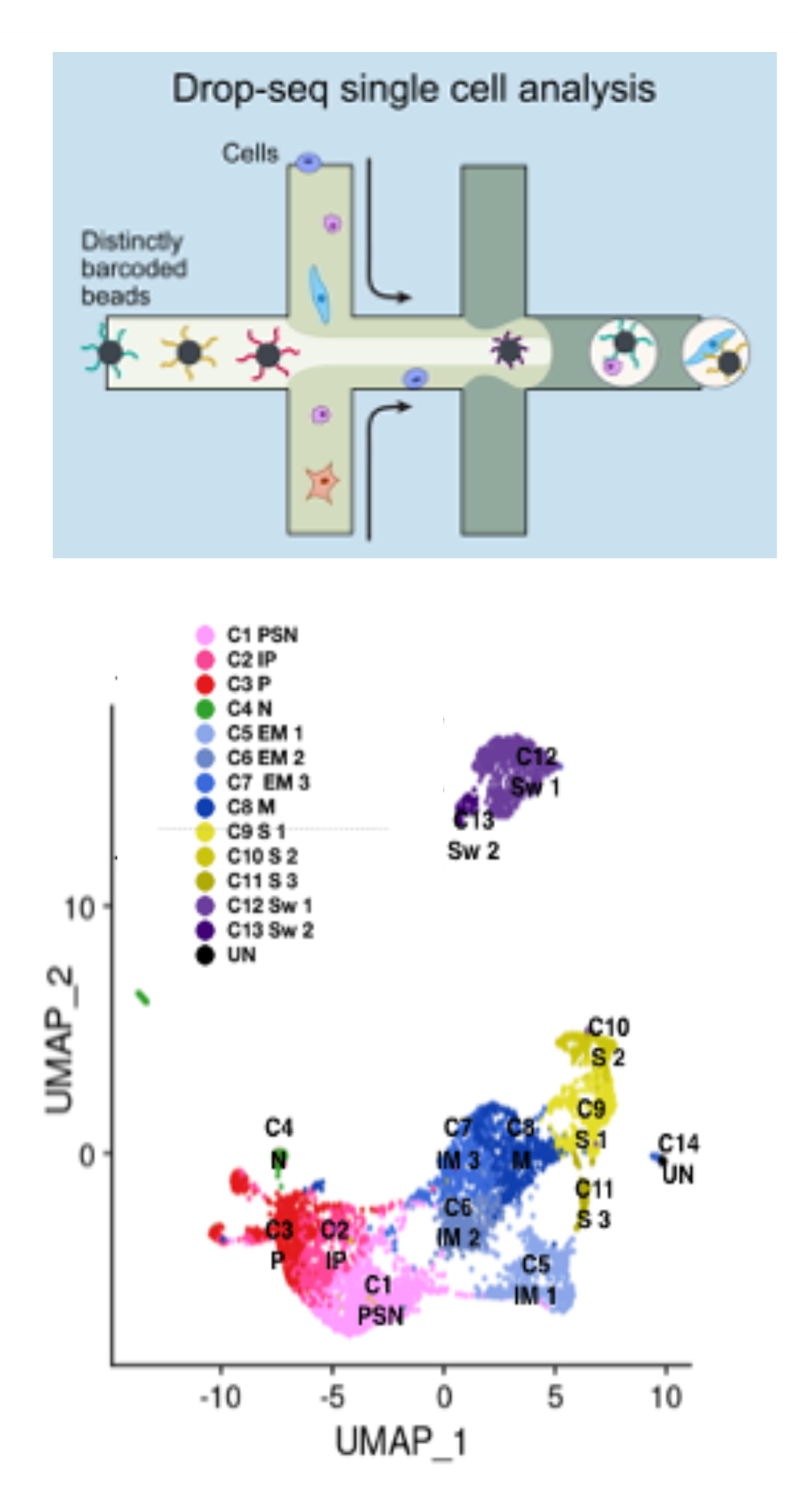 At CGS, we perform high-throughput single-cell profiling using the 10x Genomics system: 10x Genomics’s Chromium technology encapsulates every single cell inside a droplet, each equipped with a unique barcode bead (GEM, for Gel Bead-In Emulsion). This permits to pool all molecules in a single library and to resolve each cell’s identity using the cellular barcode (CB) assigned to each molecule, thus dramatically reducing library preparation costs.
At CGS, we perform high-throughput single-cell profiling using the 10x Genomics system: 10x Genomics’s Chromium technology encapsulates every single cell inside a droplet, each equipped with a unique barcode bead (GEM, for Gel Bead-In Emulsion). This permits to pool all molecules in a single library and to resolve each cell’s identity using the cellular barcode (CB) assigned to each molecule, thus dramatically reducing library preparation costs.
TRANSCRIPTOMICS AND EPIGENOMICS We implement transcriptome and DNA accessibility profiling experiments at single cell resolution (scRNA-Seq and scATAC-Seq). Recently, we also implemented joint accessibility and gene expression profiling (Multiome). ScRNA-Seq is performed routinely and allows to capture gene expression information with high accuracy. Upon reverse transcription, a molecule-specific barcode (UMI, for Unique Molecular Identifier) is annealed to each transcript molecule. This overcomes PCR amplification bias and allows precise estimation of transcripts abundance.
GENETIC PERTURBATIONS Single-cell CRISPR-based technologies (Perturb-Seq, CROP-Seq) apply scRNA-Seq on pooled genetic perturbation screens, allowing to study the effect of genetic perturbations on gene expression at single cell resolution. Gene-associated guide barcodes (GBC) are captured together with cellular tanscripts and associated to each specific cell. In the non-targeting version, the use of inheritable barcodes find applications in cell tracking experiments.
SAMPLE MULTIPLEXING Sample multiplexing with lipid-tagged indices (MULTI-Seq) enable pooling cells with sample-specific barcodes, which are captured by scRNA-Seq. This multiplexing strategy allows to pool samples in a single scRNA-Seq library, thus reducing the costs of complex experimental designs. MULTI-Seq can be possibly combined with Perturb-Seq.
CURRENT PROJECTS
Dynamics of drug resistance in triple-negative breast cancer at single-cell resolution Researchers involved: Francesca Nadalin, Matteo Marzi, Maria Pirra Piscazzi, Bianca Giuliani. Project lead: Francesco Nicassio